Snowflake ML의 CUDA-X 라이브러리¶
Snowflake Container Runtime의 CUDA-X 통합을 사용하여 코드를 변경하지 않고도 ML을 GPUs에 걸쳐 원활하게 확장하고 데이터 변환을 수행합니다. Snowflake는 NVIDIA의 cuML 및 cuDF 라이브러리를 런타임 환경에 통합했습니다. 이러한 통합을 통해 GPUs에서 scikit-learn, umap-learn 또는 hdbscan과 같은 라이브러리를 사용할 수 있습니다. 새로운 프레임워크를 배우거나 복잡한 종속성을 처리할 필요가 없습니다.
데이터 크기나 알고리즘 복잡성을 그대로 유지하면서 항목 모델링, 유전체학, 패턴 인식과 같은 복잡한 처리를 실행할 수 있습니다. 처리 시간을 줄이면 모델을 추가로 반복할 수 있습니다.
CUDA-X 라이브러리와의 통합으로 Snowflake ML Container Runtime에서 대규모 데이터 세트의 GPU 가속 처리를 지원합니다. Container Runtime을 단독으로 사용하는 것보다 처리 속도가 훨씬 더 빠를 수 있습니다.
데이터 과학을 위한 NVIDIA CUDA-X 라이브러리¶
cuML 및 cuDF와 같은 오픈 소스 라이브러리는 보다 효율적이고 확장 가능한 데이터 워크플로를 위해 GPUs를 활용합니다. 이러한 라이브러리를 사용하여 수십억 개의 행과 수백만 개의 차원이 있는 데이터를 처리할 수 있습니다. 이러한 라이브러리에 대한 자세한 내용은 `NVIDIA CUDA-X 데이터 과학<https://developer.nvidia.com/topics/ai/data-science/cuda-x-data-science-libraries>`_ 섹션을 참조하세요.
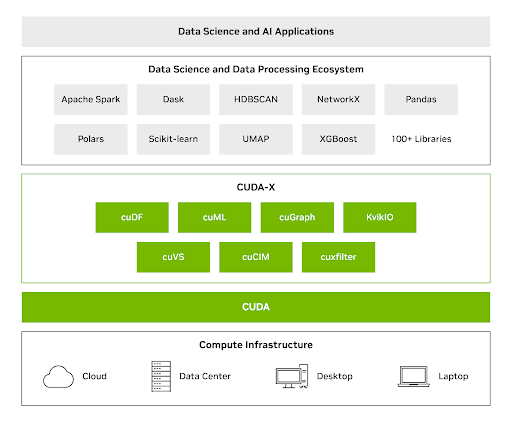
CUDA-X DS 라이브러리는 GPUs의 강력한 기능을 데이터 분석, 머신 러닝, 그래프 분석을 위해 일반적으로 사용되는 Python 라이브러리와 결합하여 팀에서 코드를 다시 작성하지 않고도 속도를 크게 향상할 수 있도록 지원합니다. CUDA-X DS를 사용하면 GPU 속도 향상을 통해 단일 GPU로 최대 테라바이트 크기의 데이터 세트를 처리할 수 있습니다.
NVIDIA cuML은 CPU 워크플로에 비해 다음과 같은 성능 개선 사항을 제공할 수 있습니다.
scikit-learn의 경우 최대 50배
UMAP의 경우 최대 60배
HDBSCAN의 경우 최대 175배
사용 사례¶
Snowflake ML Container Runtime에서의 CUDA-X 라이브러리 통합은 다음과 같은 사용 사례를 위해 Scikit-learn 및 pandas와 GPUs를 사용합니다.
대규모 항목 모델링¶
피처가 풍부한 대규모 데이터 세트에 대한 항목 모델링에는 다음이 필요합니다.
임베딩 모델 사용하기
규모에 맞게 차원 축소 적용하기
클러스터링 및 시각화를 사용하여 정확하고 관련성 높은 항목 추출하기
GPU 병렬 처리는 이전 워크플로를 더 효율적으로 수행하는 데 도움이 될 수 있습니다. cuML로 처리를 가속화하여 기존 Python 코드를 수정하지 않고도 수백만 개의 제품 리뷰를 원시 텍스트에서 잘 정의된 항목 클러스터로 변환하며, CPU에서 몇 시간씩 걸리던 작업을 GPU에서 몇 분으로 단축할 수 있습니다. 이는 UMAP 및 HDBSCAN 라이브러리에 대한 원활한 드롭인 가속화를 강조합니다.
Snowflake에서 GPUs에 대한 항목 모델링을 수행하는 방법에 대한 자세한 내용은 https://quickstarts.snowflake.com/guide/accelerate-topic-modeling-with-gpus-in-snowflake-ml/#0 섹션을 참조하세요.
컴퓨팅 유전체학 워크플로¶
Snowflake의 CUDA-X 통합을 통해 생물학적 시퀀스 처리를 크게 가속화합니다. 유전자 패밀리 예측과 같은 확장 가능한 분류 작업을 위해 DNA 시퀀스를 기능 벡터로 변환할 수 있습니다.
GPUs에서 cuDF 및 cuML을 통해 pandas와 scikit-learn 코드를 직접 실행하면 데이터 로딩, 전처리 및 앙상블 모델 학습 속도가 향상됩니다. 코드를 변경하지 않고도 기존 워크플로에 대한 GPU 가속화를 통해 연구진은 하위 수준 GPU 프로그래밍보다 생물학적 인사이트 및 모델 설계를 우선시할 수 있습니다.
Snowflake에서 개발하기¶
CUDA-X 라이브러리를 사용하여 Snowflake ML Container Runtime 내에서 GPU 가속 머신 러닝 모델을 개발하고 배포합니다. 이 섹션에서는 이러한 도구를 Python 워크플로에 통합하기 위한 단계별 가이드를 제공합니다.
시작하려면 다음을 수행합니다.
Snowflake 노트북 또는 ML 작업에서 Python 스크립트 정의하기
노트북 또는 ML 작업에 GPU 런타임 및 GPU 컴퓨팅 풀 선택하기
이전 단계를 완료한 후 다음 코드를 실행하여 사용자 환경에 CUDA-X 액셀러레이터를 구성합니다.
이제 GPUs에서 pandas 작업을 직접 실행하거나 scikit-learn, umap 또는 hdbscan 모델을 맞출 수 있습니다(GPUs에서 실행하기 위해 코드를 변경할 필요 없음). 이 예제에서는 대규모 데이터 세트에서 :code:`hdbscan`을 사용하는 방법을 보여줍니다.
적용된 사용 사례: 대규모 항목 모델링¶
계산 효율성은 대규모 텍스트 분석 및 항목 모델링에 매우 중요합니다. GPUs는 병렬 처리를 사용하여 처리 시간을 몇 시간에서 몇 분으로 단축합니다. 이 섹션에서는 CUDA-X를 사용한 GPU 가속을 통해 200,000건의 미용 제품 리뷰 데이터 세트에서 ML 모델을 가속화하는 방법을 보여줍니다.
CUDA-X를 사용하여 다음을 수행할 수 있습니다.
머신 러닝을 위해 원시 텍스트를 숫자 표현(임베딩)으로 변환합니다.
차원 축소 가속화
CUDA 라이브러리를 활용하려면 코드 시작 부분에 %load_ext cuml.accel을 추가합니다. 이를 통해 처리 시간이 몇 시간에서 몇 분으로 단축됩니다.
다음 예제 코드에서는 SentenceTransformer 클래스를 사용하여 임베딩을 생성합니다.
다음 예제 코드에서는 HDBSCAN을 사용하여 고차원 데이터를 줄입니다. 클러스터 항목을 유지합니다.
적용된 사용 사례: 복잡한 유전체학 워크플로 실행하기¶
paralog와 ortholog를 포함하는 유전자 패밀리 구성은 유전자 진화, 기능, 보존된 생물학적 과정을 이해하는 데 매우 중요합니다.
CUDA-X 라이브러리를 사용하면 분류 모델을 생성하여 DNA 시퀀스에서 유전자 패밀리를 예측할 수 있습니다. 이 모델은 게놈 주석을 가속화하고, 새로운 유전자 기능을 식별하며, 진화 경로에 대한 인사이트를 제공할 수 있습니다.
`데이터 세트<https://raw.githubusercontent.com/nageshsinghc4/DNA-Sequence-Machine-learning/master/human_data.txt>`_에는 일련의 일반 텍스트 뉴클레오티드 시퀀스와 해당 유전자 패밀리 클래스 레이블이 있습니다. 클래스는 7개의 고유한 인간 유전자 패밀리에 해당합니다.
다음 코드는 Hugging Face의 **뉴클레오타이드 변환기**를 사용하여 DNA 시퀀스를 벡터로 변환합니다. 변환기는 시퀀스를 토큰화하고 일괄 처리하여 각 유전자 시퀀스를 1280개의 기능 벡터로 변환합니다.
다음 코드를 사용하여 두 앙상블 분류 모델을 평가할 수 있습니다.
무작위 포레스트 분류기
XGBoost 분류기