CUDA-X-Bibliotheken in Snowflake ML¶
Verwenden Sie CUDA-X-Integrationen von Snowflake Container Runtime zur nahtlosen Skalierung von Datentransformationen und ML über GPUs hinweg, ohne Ihren Code ändern zu müssen. Snowflake hat cuML und cuDF-Bibliotheken von NVIDIA in die Laufzeitumgebung integriert. Mit dieser Integration können Sie Bibliotheken wie scikit-learn, umap-learn oder hdbscan mit Ihren GPUs verwenden. Sie müssen keine neuen Frameworks erlernen oder komplexe Abhängigkeiten verarbeiten.
Sie können komplexe Verarbeitungen wie Themenmodellierung, Genomik und Mustererkennung ausführen, ohne Kompromisse bei Datengrößen oder algorithmischer Komplexität einzugehen. Die Verkürzung der Verarbeitungszeit gibt Ihnen die Möglichkeit, Ihre Modelle weiter zu iterieren.
Die Integration mit den CUDA-X-Bibliotheken ermöglicht die GPU-beschleunigte Verarbeitung großer Datensets in der Snowflake ML Container Runtime. Die Verarbeitungsgeschwindigkeit kann wesentlich höher sein als bei ausschließlicher Verwendung der Container Runtime.
NVIDIA CUDA-X-Bibliotheken für Data Science¶
Open-Source-Bibliotheken wie cuML und cuDF nutzen GPUs für effizientere und skalierbarere Daten-Workflows. Sie können diese Bibliotheken verwenden, um Daten mit Milliarden von Zeilen und Millionen von Dimensionen zu verarbeiten. Weitere Informationen zu diesen Bibliotheken finden Sie unter NVIDIA CUDA-X Data Science.
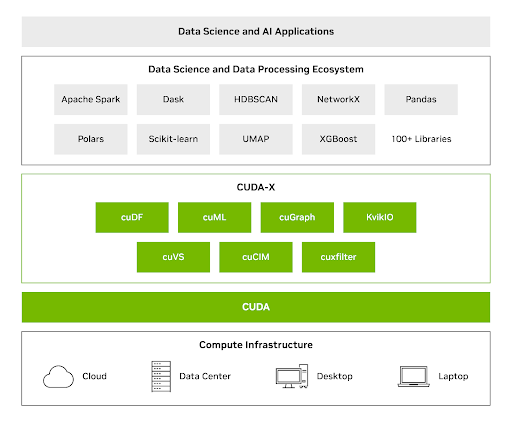
CUDA-X DS-Bibliotheken vereinen die Leistungsfähigkeit von GPUs mit häufig verwendeten Python-Bibliotheken für Datenanalyse, maschinelles Lernen und Task-Graph-Analysen – und ermöglichen so erhebliche Geschwindigkeitssteigerungen, ohne dass Teams ihren Code neu schreiben müssen. Mit CUDA-X DS können Sie die erhöhte GPU-Geschwindigkeit nutzen, um Datensets mit einer Größe von bis zu Terabytes mit einer einzigen GPU zu verarbeiten.
NVIDIA cuML kann die folgenden Leistungsverbesserungen gegenüber reinen CPU-Workflow erzielen:
Bis zu 50 x für scikit-learn
Bis zu 60 x für UMAP
Bis zu 175 x für HDBSCAN
Anwendungsfälle¶
Die Integration der CUDA-X-Bibliotheken in der Snowflake ML Container Runtime verwendet GPUs mit scikit-learn und pandas für die folgenden Anwendungsfälle:
Umfassende Themenmodellierung¶
Die Themenmodellierung für große und Feature-reiche Datensets erfordert Folgendes:
Verwenden von Einbettungsmodellen
Anwenden der Dimensionsreduktion in großem Maßstab
Verwenden von Clustering und Visualisierung, um genaue und relevante Themen zu extrahieren
GPU-Parallelität kann Ihnen helfen, die vorhergehenden Workflows effizienter zu gestalten. Indem Sie Ihre Verarbeitung mit cuML beschleunigen, können Sie Millionen von Produktbewertungen aus Rohtext in genau definierte Themencluster umwandeln, die sich von Stunden auf CPUs bis Minuten auf GPUs ohne Änderungen an bestehendem Python-Code reduzieren lassen. Dies unterstrich die nahtlose Drop-in-Beschleunigung für UMAP- und HDBSCAN-Bibliotheken.
Weitere Informationen zur Durchführung der Themenmodellierung über GPUs auf Snowflake finden Sie unter https://quickstarts.snowflake.com/guide/accelerate-topic-modeling-with-gpus-in-snowflake-ml/#0.
Rechengestützte Genomik-Workflows¶
Verwenden Sie die CUDA-X-Integrationen von Snowflake zur deutlichen Beschleunigung der Verarbeitung von logischen Sequenzen. Sie können DNA-Sequenzen in Feature-Vektoren für skalierbare Klassifizierungsaufgaben, wie z. B. die Vorhersage von Genfamilien, konvertieren.
Das Ausführen von pandas- und scikit-learn-Code direkt auf GPUs mit cuDF und cuML beschleunigt das Laden von Daten, die Vorverarbeitung und das Training von Ensemble-Modellen. Diese GPU-Beschleunigung für bestehende Workflows ohne Code-Änderungen ermöglicht es Forschenden, logischen Erkenntnissen und Modelldesign Vorrang vor einfacherGPU-Programmierung zu geben.
Entwickeln in Snowflake¶
Verwenden Sie die CUDA-X-Bibliotheken zum Entwickeln und Bereitstellen von GPU-beschleunigten Machine-Learning-Modellen innerhalb der Snowflake ML Container Runtime Dieser Abschnitt bietet eine Schritt-für-Schritt-Anleitung für die Integration dieser Tools in Ihre Python-Workflows.
Um zu beginnen, gehen Sie wie folgt vor:
Definieren Sie Ihr Python-Skript in einem Snowflake Notebook oder einem ML-Job.
Wählen Sie den die GPU-Laufzeit und einen GPU-Computepool für Ihr Notebook oder Ihren ML-Job.
Nachdem Sie die vorherigen Schritte ausgeführt haben, führen Sie den folgenden Code aus, um die CUDA-X-Beschleuniger in Ihrer Umgebung zu konfigurieren.
Jetzt können Sie pandas-Operationen direkt über GPUs ausführen oder das scikit-learn-, umap- oder hdbscan-Modell anpassen (beachten Sie, dass keine Codeänderung für die Ausführung über GPUs erforderlich ist). In diesem Beispiel wird die Verwendung von hdbscan für große Datensets veranschaulicht:
Anwendender Anwendungsfall: Themenmodellierung in großem Maßstab¶
Die Recheneffizienz ist entscheidend für umfangreiche Textanalysen und Themenmodellierung. GPUs verwenden die Parallelverarbeitung, um die Verarbeitungszeit von Stunden auf Minuten zu reduzieren. In diesem Abschnitt erfahren Sie, wie Sie ML-Modelle für ein Datenset von 200.000 Bewertungen von Pflegeprodukten mithilfe der GPU-Beschleunigung mit CUDA-X beschleunigen können.
Mit CUDA-X können Sie Folgendes tun:
Umwandeln von Rohtext in numerische Darstellungen (Einbettungen) für maschinelles Lernen.
Beschleunigen der Dimensionsreduktion
Um die CUDA-Bibliotheken zu nutzen, fügen Sie %load_ext cuml.accel am Anfang Ihres Codes hinzu. Dies reduziert Ihre Bearbeitungszeit von Stunden auf Minuten.
Der folgende Beispielcode verwendet die SentenceTransformer-Klasse, um Einbettungen zu erstellen.
Der folgende Beispielcode verwendet HDBSCAN zur Reduzierung hochdimensionaler Daten. Die Clusterthemen bleiben erhalten.
Anwendender Anwendungsfall: Ausführen komplexer Genomik-Workflows¶
Die Organisation von Genfamilien, die Paralogien und Orthologien umfasst, ist entscheidend für das Verständnis der Genentwicklung, der Genfunktion und konservierter biologischer Prozesse.
Mit den CUDA-X-Bibliotheken können Sie ein Klassifizierungsmodell erstellen, um Genfamilien anhand von DNA-Sequenzen vorherzusagen. Dieses Modell kann die genomische Annotation beschleunigen, neuartige Genfunktionen identifizieren und Einblicke in evolutionäre Pfade liefern.
Das Datenset enthält eine Reihe von Nukleotidsequenzen in Klartext und die entsprechenden Genfamilienklassenbezeichnungen. Die Klassen entsprechen sieben verschiedenen menschlichen Genfamilien.
Der folgende Code verwendet den Nukleotid-Transformator von Hugging Face, um die DNA-Sequenzen in Vektoren umzuwandeln. Der Transformer tokenisiert und gruppiert die Sequenzen, um jede Gensequenz in einen 1280-Feature-Vektor umzuwandeln.
Sie können den folgenden Code verwenden, um zwei Ensemble-Klassifikationsmodelle zu bewerten:
Ein Random-Forest-Klassifikator
Ein XGBoost-Klassifikator