Snowflake ML のCUDA-Xライブラリ¶
Snowflake Container Runtimeの CUDA-X 統合を使用して、コードを変更せずに GPUs でデータ変換と ML をシームレスにスケールします。Snowflakeは NVIDIA の cuML および cuDF ライブラリをランタイム環境に統合しました。 この統合により、scikit-learn、umap-learn、hdbscanなどのライブラリを、 GPUs で使用することができます。新しいフレームワークを学習したり、複雑な依存関係を処理したりする必要はありません。
データサイズやアルゴリズムの複雑さを妥協することなく、トピックモデリング、ゲノミクス、パターン認識などの複雑な処理を実行できます。処理時間を短縮することで、モデルでさらに反復する機会が得られます。
CUDA-Xライブラリとの統合により、Snowflake ML Container Runtimeにおける大規模なデータセットの処理を GPU で高速化できます。 Container Runtimeのみを使用するよりも桁違いに処理速度が速くなる可能性があります。
データサイエンス用の NVIDIA CUDA-Xライブラリ¶
cuML や cuDF のようなオープンソースライブラリは、 GPUs を利用して効率的でスケーラブルなデータワークフローを実現します。これらのライブラリを使用して、数十億の行と数百万のディメンションを持つデータを処理できます。これらのライブラリの詳細については、 NVIDIACUDA-Xデータサイエンス をご覧ください。
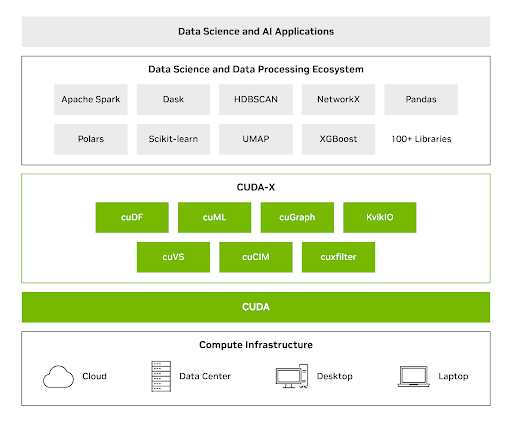
CUDA-X DS ライブラリは、 GPUs のパワーと、データアナリティクス、機械学習、グラフ分析に一般的に使用されるPythonライブラリを組み合わせ、チームがコードを書き換えることなく、大幅なスピードアップを実現します。CUDA-X DS を使用すると、 GPU の速度向上により、単一の GPU で最大でテラバイトのデータセットを処理できます。
NVIDIA cuML は CPU ワークフローで以下のパフォーマンス向上を実現できます。
scikit-learnの場合、最大50倍
UMAP の場合、最大60倍
HDBSCAN の場合、最大175倍
ユースケース¶
Snowflake ML Container Runtimeにおける CUDA-Xライブラリの統合は、以下のユースケースに GPUs とScikit-learnとpandasを使用します。
大規模なトピックモデリング¶
大規模で特徴量が豊富なデータセットのトピックモデリングには、次が必要です。
埋め込みモデルの使用
大規模な次元削減の適用
クラスタリングと視覚化を使用した、正確かつ関連性のあるトピックの抽出
GPU 並列処理により、前述のワークフローをより効率的に達成することができます。cuML で処理を加速させることで、数百万件の製品レビューをrawテキストから明確に定義されたトピッククラスターに変換することができ、既存のPythonコードを変更することなく、 CPU の数時間を GPU での数分に削減できます。これは、 UMAP および HDBSCAN ライブラリのシームレスなドロップインアクセラレーションを強調しています。
Snowflakeの GPUs でのトピックモデリング実行に関する詳細は、 https://quickstarts.snowflake.com/guide/accelerate-topic-modeling-with-gpus-in-snowflake-ml/#0 をご参照ください。
計算ゲノミクスワークフロー¶
Snowflakeの CUDA-X統合を使用して、生物学的な配列の処理を大幅に高速化します。DNA 配列を、遺伝子ファミリーの予測など、スケーラブルな分類タスクの特徴ベクトルに変換できます。
cuDF および cuML を使用してpandasとscikit-learnのコードを GPUs で直接実行すると、データのロード、前処理、アンサンブルモデルトレーニングを高速化できます。コードを変更することなく、既存のワークフローを GPU で高速化することで、研究者は下位レベルの GPU プログラミングよりも生物学的な洞察と設計を優先することができます。
Snowflakeでの開発¶
CUDA-Xライブラリを使用して、Snowflake ML Container Runtime内で GPU で加速させた機械学習モデルを開発しデプロイします。このセクションでは、これらのツールをPythonワークフローに統合するための手順ガイドを提供します。
開始するには、以下の手順を実行します。
Snowflake Notebookまたは ML ジョブでPythonスクリプトを定義する
Notebookまたは ML ジョブ用の GPU ランタイムと GPU コンピューティングプールを選択する
前述の手順を実行したら、次のコードを実行して環境内で CUDA-Xアクセラレーターを構成します。
これで GPUs でpandas操作を直接実行できるようになりました。または、scikit-learn、umap、hdbscanモデルを適合させることができます( GPUs で実行するためにコードを変更する必要はありません)。この例は、大規模なデータセットで hdbscan を使用する方法を示しています。
適用されるユースケース:大規模なトピックモデリング¶
計算効率は大規模なテキスト分析とトピックモデリングに不可欠です。 GPUs は並列処理を使用して、処理時間を数時間から数分に短縮します。このセクションでは、 GPU による高速化と CUDA-Xを使用して、200,000件の美容製品のレビューのデータセットに対して ML モデルを高速化する方法を示しています。
CUDA-Xを使用して次を実行できます。
機械学習のために、rawテキストを数値表現(埋め込み)に変換する
次元削減を加速化する
CUDA ライブラリを利用するには、コードの先頭に%load_ext cuml.accelを追加します。これにより、処理時間が数時間から数分に短縮されます。
次のコード例では、 SentenceTransformer クラスを使用して埋め込みを作成しています。
次のコード例では、 HDBSCAN を使用して高次元データを削減しています。クラスタートピックを保持します。
適用されるユースケース:複雑なゲノミクスワークフローの実行¶
パラログとオーサログを含む遺伝子ファミリー組織は、遺伝子の進化、機能、および生物学的プロセスを理解するために重要です。
CUDA-Xライブラリを使用すると、 DNA 配列から遺伝子ファミリーを予測する分類モデルを作成できます。このモデルは、ゲノムアノテーションを加速させ、新しい遺伝子機能を特定し、進化経路に関する洞察を提供することができます。
データセット には、一連のプレーンテキストのヌクレオチド配列と、それに対応する遺伝子ファミリークラスラベルがあります。クラスは、7つの異なるヒトの遺伝子ファミリーに対応します。
次のコードは、Hugging Faceの ヌクレオチドトランスフォーマー を使用して、 DNA 配列をベクトルに変換します。トランスフォーマーは、各遺伝子配列を1280の特徴ベクトルに変換するために、配列をトークン化してバッチ処理します。
次のコードを使用して、2つのアンサンブル分類モデルを評価できます。
ランダムフォレスト分類子
XGBoost 分類子